Table of Contents
Abstract
Manuelito allows a rapid interpretation of complex posttranslational modification patterns based on the peptide mass fingerprints of proteins. It can be applied to proteins that contain natural PTMs as well as modifications introduced after isolation of the polypeptides.
The software package comprises a Java 2 standalone application that can be run on every Java-enabled platform and a Java web archive to be run as web server.
Posttranslational modifications (PTMs) are important modifiers of the physiological functions and behaviours of proteins like localization, intermolecular interactions, and enzymatic activities. Prime example for modulation of enzyme activities is the phosphorylation of enzymes of the metabolic pathway. After translation of the target protein, highly specific enzymes covalently link activated molecules to specific amino acid residues. In case of phosphorylation, ATP is the activated donor of phosphate and kinases are the enzymes that attach the phosphates to the proteins. Amino acids with free hydroxyl groups are usually targets for phosphorylation, i.e serines, threonines, tyrosines with serines being most frequently phosphorylated. The covalent linking of a chemical group is not a dead end street: there are also specific enzymes that can remove the modifications (e.g phosphatases that remove phosphates). Such a removal will, of course, reset the physiological behavior of the protein.
To summarize, the PTM framework comprises target proteins, activated substrates that provide the chemical group to be added, specific enzymes that transfer specific groups to specific residues of specific proteins, and specific enzymes that can again remove these groups. PTMs provide a means to reversibly change behaviours of proteins in a highly selective manner, and, actually, this feature is exploited extensively by the cells.
Many proteins contain more than a single residue that can be potential target for a PTM. In fact, many proteins appear to be multiply modified. Multiple modifications can not only serve as simple potentiator of a single modification. The simultaneous placement of different modifications might provide an additional level of coding a certain physiological function. Examples are nuclear factors that are potential acceptors for e.g. phosphorylation, acetylation and methylation groups. In the case of histone proteins, a clear coding potential by these modifications has been postulated: it is believed that combinations of certain modifications can be translated into a certain gene expression level.
Specific modifications of proteins are usually detected by protein- and modification- specific antibodies. This approach is based on raising antibodies against known modifications on known target sites. Two major drawbacks characterize this procedure: first, antibodies can only be raised against known target and secondly, the binding of an antibody can be compromised by an adjacent modification. An unbiased detection therefore requires a different technology.
Mass Spectrometry (MS) is a analysis technique to determine masses of molecules. Depending on the kind of instrument, molecules are ionized and released into an electric field where they are separated according to charge and mass. The primary strength of MS is precision. Masses can be determined with a precision of less than 5 ppm.
Due to their big mass, proteins are usually analyzed as small fragments (peptides) that are obtained by digesting the proteins with a highly specific protease. The masses of the peptides derived from one protein are its mass fingerprint, i.e. a list of mass values that is highly specific for a protein. This peptide mass fingerprint (PMF) can therefore be used to identify an unknown protein. The instrument that is used to get PMFs of proteins in one-dimensional MS is e.g. the MALDI (Matrix Assisted Laser Desorption Ionisation) mass spectrometer.
In order to get detailed information (e.g. the amino acid sequence) about the composition, the peptide can be fragmented even further within MS/MS instruments. The released fragments can give hints about the basic elements (amino acids) of the parent peptide.
PTMs add additional mass to proteins and peptides. With masses of modifications and native peptides being known, MALDI-MS can give information about whether a peptide carries which PTMs. However, positional information usually can't be obtained, i.e.it can't be determined on which residue the modification is placed in case there are more target sites than modifications attached.
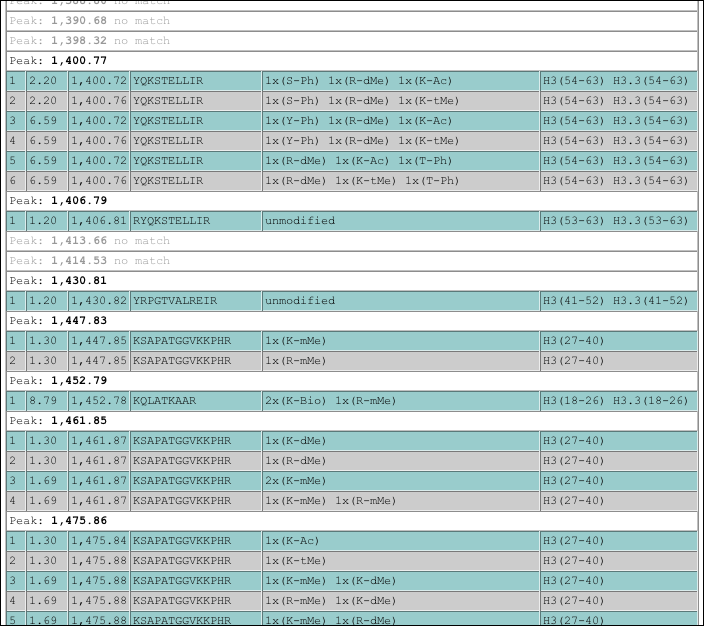
It is evident that MALDI-MS cannot uncover positional information but can give first hints on the type and number of modifications present. In mathematical terms it decodes repetitive combinations NOT permutations.
For positional information MS/MS comes into play. By fragmenting the modified peptide, mass shifts of single amino acids can be visualized. This leads to identification of the modification's target residue. Unfortunately MS/MS procedures are tedious to perform and precise information is not always obtained.
The biological question behind the analysis of modification patterns is whether specific modification states correlate with specific physiological states. A strategy to efficiently get a comprehensive picture in various situations requires rapid and systematic analyses of different specimen. Therefore a screening procedure is desired that can limit the workload on bottleneck applications. Even though MALDI-MS has the aforementioned limitations, it can serve a useful function even in decrypting complex PTM patterns: it can be used to screen for changes in masses that could be indicative for changes in modification states. The peptides giving rise to changes can then be sequenced by MS/MS, the bottleneck application, and the precise position of modifications can be mapped.
Such a strategy requires that spectrum peaks (the masses) are being properly assigned to peptides and their modification states. Interpretation spectra is the job of computers and it is on the software to do the proper assignments.
Manuelito's task is to consider all modification states of peptides when trying to match peaks. In addition, Manuelito can handle modification events that cause selective modifications of residues already modified
Rational behind this feature is a particular way to process the peptides for analysis. Several procedures require the peptides to be chemically derivatized. This derivatization is basically a modification reaction that is applied to the peptide before MS. As it is for the naturally occurring modifications, the synthetic ones added by the researcher have a residue specificity and a specific mass. Reason for such a treatment are for example quantification experiments in which two different samples are analyzed at the same time. This procedure based on two different processing procedures for the two samples, i.e. 2 different chemical derivatization reactions. Another example is given by the analysis of histone proteins. These proteins are highly charged and therefore difficult to efficiently 'shoot' in the MS instrument. With chemical compounds that specifically target highly charged residues (i.e. lysines) the peptides charge can be neutralized.
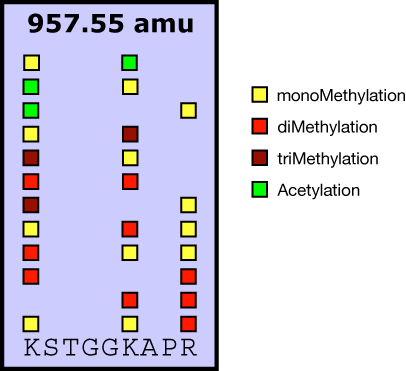
Figure 2. Selective Chemical Modification
Same example: if the same proteins as in figure 1 were acetylated chemically (heavy acetyl e.g.) after protease cleavage, a diversification of masses would occur. In addition to all peptides getting acetylated at the N-terminus, unmodified and monomethylated lysines would also get an additional acetyl group.
Two important features characterize chemical modification reactions: a) these reactions can be considered obligatory, they will happen at almost 100% efficiency on each target residue b) but the linking of a chemical group might be excluded by an already existing natural modification.
Manuelito is the only software that can consider this modification-specific exclusivities and is therefore the only software the can be used to completely interpret MALDI-MS spectra of peptides that have been subjected to chemicals that exhibit such a behaviour.
In summary, Manuelito will generate protease cleavage fragments of a given set of proteins. For each peptide generated it will iterate through all possible modification states to try to match it to one of the peaks that have been passed to the application.
One or more proteins are the source of the peptides analyzed by MALDI-MS.
These proteins were target of various modification events (outside of labs, i.e. in nature). These modifications are considered variable.
Optionally, the proteins are now or after the next step subjected to one further modification cycle. This is now in-lab modification, referred to as chemical modification. Chemical Modifications are considered to happen at 100% efficiency on unmodified target residues and, according to defined rules, on naturally modified ones as well.
Proteins are subjected to protease cleavage that will not be affected by any modification (this is mainly due to lack of rules).
The cleavage fragments are analyzed by mass spectrometry and give rise to peaks in the MS spectrum.
The MS operator parses the peaks for obvious contaminations (keratins etc.).
All masses are considered to be monoisotopic masses.
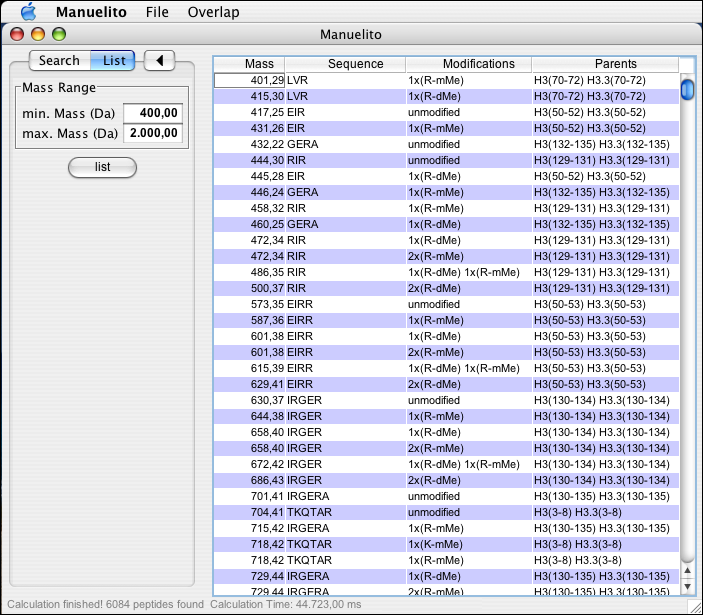
Manuelito performs a match of the peak masses to theoretical peptides. As Manuelito considers virtually all modification scenarios per peptide, the efficiency, i.e. speed, of operation depends on the number of variable ('natural') modifications the user selects and the number of target residues within the cleavage fragments. It is therefore evident that the peptides should be fairly small, i.e. the user should limit the mass range of peak masses supplied. We suggest a range from 400-2500 Da. It is anyway almost impossible to further analyze larger peptides (by MS/MS for example).
Based on the number and kind of modifications that are present on a matched peptide, as well as the number of missed cleavages that generated the peptide, Manuelito will assign a quite arbitrary penalty score to the match.
Manuelito expects the user to provider a list of peptides to work on. Detailed analysis of a protein's modification state requires a biochemical purification before MS. Ideally, only few, ideally only one, protein in fairly high purity should be the starting point as too many contaminants might lead to false positives.
The standalone application allows for entering protein sequences manually. Preferred, and the only option in the web interface is input via loading of FASTA formatted files. Protein sequences will always be parsed for illegal amino acids (legal are unambiguous amino acids single-letter IUPAC code).
The user can select from a set of defined naturally occurring modifications the ones that he expects to be present the proteins he analyzed. Natural Modifications are variable modifications (MASCOT term), i.e. Manuelito keeps considering unmodified target residues.
All natural modifications are excluded by preexisting modifications.
Table 1. Natural Modifications
| modification | target residue | monoisotopic mass |
|---|---|---|
| acetylation | lysine | 42.010565 |
| monomethylation | lysine, arginine | 28.0313 |
| dimethylation | lysine, arginine | 14.01565 |
| trimethylation | lysine | 42.04695 |
| phosphorylation | serine, threonine, tyrosine | 79.966331 |
| biotinylation | lysine | 226.0776 |
The user can select from a list of provided protease. The proteases are considered not to be influenced by modifications on or close to their target residues.
Depending on the proteases, the efficiency of cleavage varies and there might be 'missed' cleavages on the proteins. The number of missed cleavages defines the maximum number of missed cleavages that are allowed per generated peptide. I.e. a value of 2 will generate peptides with a maximum of 2 internal protease target residues.
Simple option to indicate that the protease cleavage was performed after the chemical modification. Reason for this parameter is the modification of free N-terminal ends by many chemical compounds. After protease treatment, a chemical modification will hit the N-termini of all fragments, while performing the derivatization before leads to the modification of solely the full protein's N-terminus.
Apart from the common chemical reactions that can happen or can be performed on cysteine and methionine residues, Manuelito can consider further chemical modifications that may server a useful function in distinguishing modification patterns.
These modifications exhibit a defined exclusivity on naturally premodified target residues. I.e. Propionylation will happen on unmodified lysines and monomethylated ones, while all other modifications will prevent further modification.
A list of masses derived from MALDI-MS runs. Ideally the peaks should be filtered for contaminants in order to reduce workload and false positives.
The tolerance that should be applied in the search. Units of tolerance are Dalton and ppm. Recommended setting: the lower, the better. A well calibrated MS spectrum allows values lower than 50 ppm, while uncalibrated spectra should be analyzed with at least 100 ppm. Please note that there will be much more false positives if the tolerance value is high (>50 ppm).
Full GUI application. Parameters are entered and results are presented through the graphical interface.
Manuelito tries to assign all possible peptides and their modification states to experimental peak masses that are derived from proteolysis and MS of a limited set of purified peptides. It will take into account chemical modification reactions that are performed either before or after proteolysis.
Two result sets can be overlapped in order to identify peptides that are very likely to be present in the specimen. This feature exploits the software's ability to consider modification-specific exclusivity of certain chemical derivatization reactions. If, e.g. the same sample is treated in parallel with the same protease but with and without certain chemicals, then possible ambiguities in the assigned modification patterns can be eliminated by overlapping the two result sets.
The software can compute all theoretical peptides with all their modification states and masses that can be derived of a limited set of proteins.
Results can be printed or exported to tab-delimited files that can be opened with e.g. Microsoft Excel.
The application state can be saved as a reusable project file.
Java 1.4.* or higher should be installed. You can get the Java Runtime Environment here.
If you are the lucky owner of an apple computer, you should also be able to raise money for OSX. In case you still run on OS 9.2 or before, you won't be able to get the required Java runtime environment.
OSX and Windows 2000+ will allow for a simple double-click driven startup. In all other cases start from commandline:
>java -jar manuelito.jar
Note in advance: You can alwas store the current settings and the peptides in the result table into a manuelito project file that can be re-opened at later time (->).
You will be presented one main window. There won't be many more. On the left side of the windows you will find 3 tabs. The main parameter input is done in the 'Settings' tab. Here the parameters for both, the search and the list output can be set.
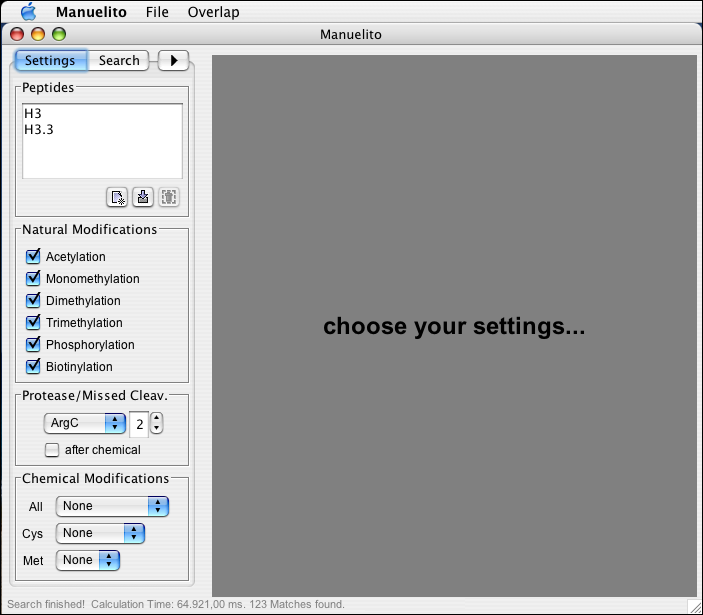
Proteins can be entered either manually or loaded as single or multiple proteins from a FASTA formatted file. The names of the proteins will appear in the list and can be selectively removed by choosing and pressing the delete button.
The protease and modifications that can be chosen are very limited at present. This is due to our specific application. In case you want to add more options, have a look the developers guide or send me an email (tobias.straub_at_lmu.de).
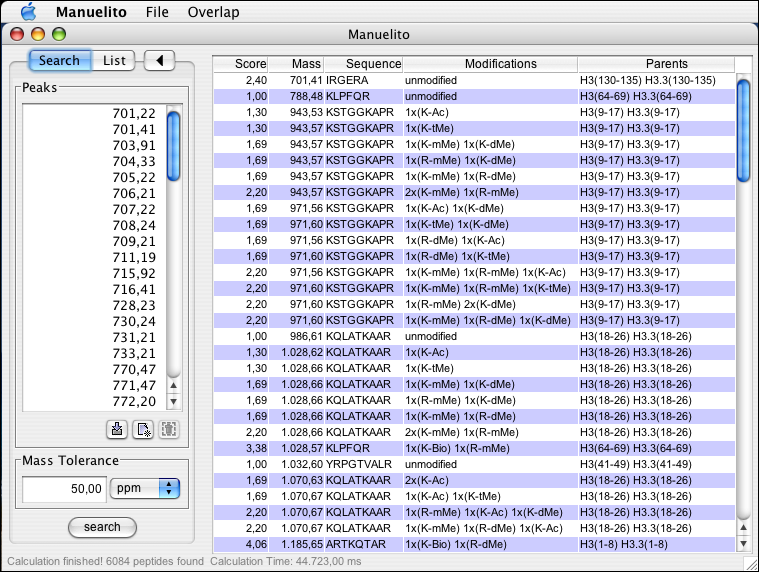
A search can be performed after providing a list of peak masses. The easiest way to get the masses into Manuelito is to copy them from your preferred MS application (or MS Excel )and paste them into Manuelito (using either the paste button or right-clicking into the list panel). Note: Manuelito interprets localized number formats, that means that your preferred copy-from application (Excel) e.g. is running the same number format as your operating system.
After the search button is pressed a progress bar will indicate the progresson of the search. Matching peptides will appear in the right table. The results can be sorted by clicking into the respective header (each click reverts the current search order). Furthermore the results can be saved into a tab-delimited file (->) that can be openend with Excel. You can also print the result table using ->
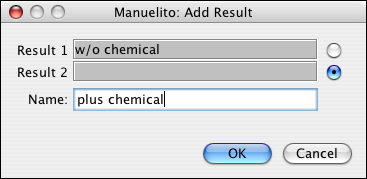
In order to overlap peptides from two different processing procedures, single results have to placed into an overlap set. This is done by calling ->. You can then enter the current result peptides into one of the two slots available.
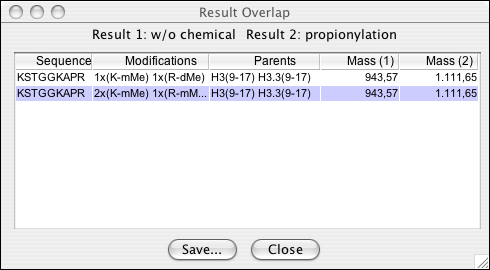
As soon as there are two result sets placed into two overlap slots, the overlap can be displayed (->) The overlapping peptides can then be saved into a tab-delimited list.
It is furthermore possible to have Manuelito compute all possible peptides and their modification states that can be derived from a given set of peptides. In the 'List' tab, a mass range for the theoretical peptides can be entered. After pressing the 'list' button, all possibilities will be listed. They can be saved and exported as the search results.
If the search of list process crashes, there is no way to recover but to quit the application and restart.
Copying and pasting mass values (peaks) from i.e. Excel into Manuelito requires the numbers to be in the operating systems number format. This can be a problem when copying from non-localized MS applications. If you fail pasting your values this is most likely the problem. In theses cases try to paste the values into a text editor and edit the numbers there to reflect your OS number format.
Saved projects are potentially incompatible with future versions.
A computer running a servlet container such as Jakarta Tomcat or JBoss. Manuelito has so far only been tested with Tomcat 5.0.
Install manuelito.war as described in the manual of your servlet engine. Manuelito.war will by default be installed in the Manuelito context.
In the web input form you can set the same paramters as in the standalone application. Proteins can only be entered by uploading a FASTA file. Peak masses can only be enter in US format (whith '.' as decimal separator).
Optional output formats comprise: HTML, Text (tab-delimited), XLS (EXCEL)
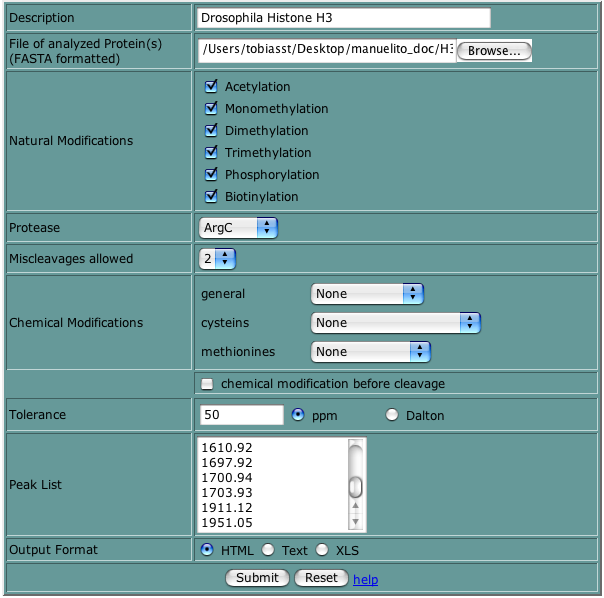
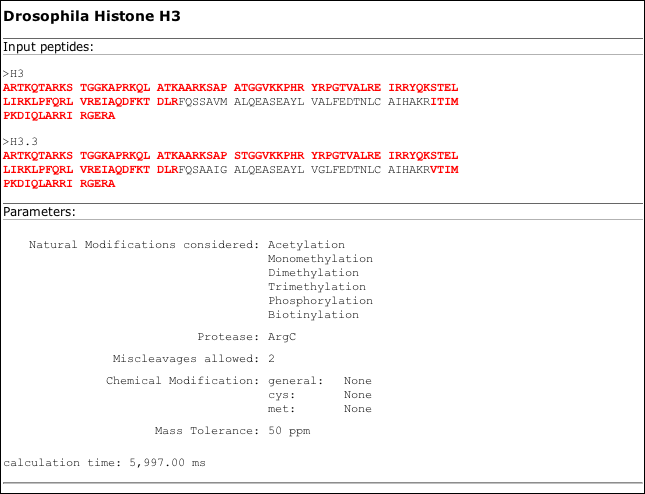
The pure HTML result output summarizes the input paramters as well as the sequence coverage of the input proteins by the result peptides.
The result table also lists the peak masses that did not pick up a match in the search.